Protein ikincil yapısı
Biyokimya ve yapısal biyolojide ikincil yapı, protein veya nükleik asit (DNA/RNA) gibi biyopolimerlerin yerel parçalarının genel, üç boyutlu biçimleridir. Buna karşın, atomlarının üç boyutlu uzaydaki konumları üçüncül yapı tanımlamasına girer.
İkincil yapının resmî tanımı, bir biyopolimerin hidrojen bağı yapıları ile tanımlanabilir; bunlar atomik çözünürlüklü bir yapı aracılığıyla ile belirlenir. Proteinler durumunda, ikincil yapı, protein omurgasındaki amit ve karboksil grupları arasındaki hidrojen bağlarının örüntüsü ile tanımlanır. Nükleik asitlerde, ikincil yapı azolu bazlar arasındaki hidrojen bağlarının örüntüsü ile tanımlanır. Bazen hidrojen bağı örüntülerinde önemli distosiyon olabilir, böyle durumlarda ikincil yapının otomatik öngörüsü zor olabilir.
Proteinlerde ikincil yapı Ramachandran grafiğininin belli bir bölgesindeki omurga dihedral açılar örüntüsüne bağlı olarak da tanımlanabilir. Yani, bu dihedral açılara sahip bir amino asit kalıntılarından oluşan bir protein parçası, sarmal (heliks) olarak adlandırılabilir, gerçekten doğru hidrojen bağlarına sahip olup olmadığına bakılmaksızın. Genelde, yapısı çözülmüş bir proteinin PDB dosyasında onun ikincil yapısı da belirtilir.
Bir biyopolimerin ikincil yapı içeriği (örneğin "bu protein %40 α-sarmal ve %20 β-yapraktır" demek gibi) çoğu zaman spektroskopik olarak kestirilebilir. Proteinler için yaygın bir yöntem, uzak mor-ötesi (170-250 nm) dairesel dikroizmdir. 208 ve 222 nm'de belirgin bir çifte minimum, α-sarmal yapıya işaret eder, buna karşın 204 nm veya 217 nm'de tek bir minimum, sırasıyla, rastgele sarım veya β-yaprak yapının işaretidir. Daha ender kullanılan bir yöntem, kizilötesi spektrodkopisidir, bununla hidrojen bağlanmasına duyarlı olan amit gruplarının osilasyonlarına bakılır. İkincil yapı içeriğini hassas şekilde kestirmek için kullanılan bir diğer yöntem de, Nükleer manyetik rezonans spektrumundaki bazı kimyasal kaymalara bakmaktır.
ikincil yapı terimi 1952'de Stanford Universitesi'nde bulunan Kaj Ulrik Linderstrøm-Lang tarafından türetilmiştir.
Protein
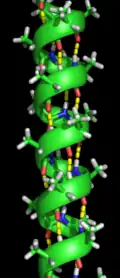
Proteinlerde ikincil yapı, amino asit kalıntıları arasındaki hidrojen bağları tarafından belirlenir. En yaygın ikincil yapılar alfa sarmal ve beta yapraktır. Başka sarmal tipleri, 310 helix ve π sarmalın da enerjetik bakımdan uygun hidrojen bağı örüntüleri olduğu hesaplanmıştır; ancak doğal proteinlerde bu tip sarmallar genelde ancak alfa-sarmalların uçlarında görülür, uygunusuz istiflenmeden dolayı ortalarda görülmezler. Poliprolin sarmal ve alfa yaprak gibi uzun veya geniş yapılar doğal proteinlerde ender bulunur ama bu yapıların protein katlanma sürecinde önemli oldukları hipotezlenir. "Normal" ikincil yapı elemanları arasında sıkı dönüşler ve gevşek, esnek ilmikler bulunur. Rastgele sarmal gerçek bir ikincil yapı elemanı değildir, düzenli bir ikincil yapının yokluğuna karşılık gelen konformasyonlar kümesidir.
Amino asitler ikincil yapı elemanları oluşturma yetenekleri bakımından çeşitlilik gösterir. Prolin ve glisin bazen "sarmal kırıcı" olarak bilinir, çünkü α-sarmal omurga yapısının düzenliliğini bozarlar. Ancak, bunların ikisinin de olağandışı özellikleri vardır ve bu yüzden dönüşlerde sık bulunurlar. Sarmal bir konformasyon edinmeyi tercih eden amino asitler arasında lizin, glutamat, metyonin, alanin ve lösin bulunur. Buna karşın, büyük aromatik kalıntılar (triptofan, tirozin ve fenilalanin) ve -dallı amino asitler (izolösin, valin, ve treonin) beta yaprak konformasyonunu tercih ederler. Ne var ki bu tercihler çok güçlü değildir, bu yüzden sadece protein dizisine dayanarak ikincil yapıyı öngörmek güvenilir değildir.

Protein ikincil yapısını tanımlamak için birkaç yöntem vardır (örneğin, DEFINE,[1] DSSP,[2] STRIDE (protein)[3]).
| Geometrik özellik | α-helix | 310 sarmalı | π-sarmal |
|---|---|---|---|
| Dönüş başına kalıntı | 3.6 | 3.0 | 4.4 |
| Kalıntı başına ötelenme | 1.5 Å (0,15 nm) | 2.0 Å (0,20 nm) | 1.1 Å (0,11 nm) |
| Sarmal yarıçapı | 2.3 Å (0,23 nm) | 1.9 Å (0,19 nm) | 2.8 Å (0,28 nm) |
| Hatve | 5.4 Å (0,54 nm) | 6.0 Å (0,60 nm) | 4.8 Å|(0,48 nm) |
DSSP kodu

Protein İkincil Yapılar Sözlüğü (İngilizce Dictionary of Protein Secondary Structure, kısaca DSSP olarak bilinir) protein ikincil yapılarını tek harfli kodlar ile betimler. İkincil yapı, hidrojen bağı örüntülerine dayanarak tayin edilir. Bu hidrojen bağı örüntüleri Pauling ve arkadaşları tarafından 1951'de, deneysel olarak herhangi bir protein yapısı daha çözülmeden, önerilmişti. DSSP sekiz tip ikincil yapı tanımlar:
- G = 3-dönmeli sarmal (310 sarmal). Asgari uzunluk 3 kalıntıdır.
- H = 4-dönmeli sarmal (α sarmal). Asgari uzunluk 4 residues.
- I = 5-dönmeli sarmal (π sarmal)). Asgari uzunluk 5 residues.
- T = hidrojen bağlı dönüş (3, 4 or 5 dönmeli)
- E = paralel veya anti-paralel β-yaprak konformasyonlu uzun şeritler. Asgari uzunluk 2 kalıntıdır.
- B = izole bir β-köprüye ait bir kalıntı (tek bir çift β-yaprak hidrojen bağı oluşumu)
- S = kıvrım (hidrojen bağına dayanmayana tek tanımlama).
Yukarıdaki konformasyonlardan birinde olmayan amino asit kalıntıları, sekizinci bir tip sayılan 'sarım' (İngilizce 'coil') olarak tasnif edilirler: çoğu zaman ' ' (boşluk), c (coil, yani sarım) veya '-' (tire). Sarmallar (G,H ve I) ve yaprak konformasyonlarının makul bir uzunluğa sahip olmak zorundadır. Bu demektir ki, birincil yapıdaki iki yan yana kalıntı aynı hidrojen bağı örüntüsünü oluşturmalıdır. Eğer sarmal ve hidrojen bağlanma örüntüsü çok kısaysa, (sırasıyla) T veya B olarak tanımlanırlar. Kalıntıları başka protein ikincil yapı kategorilerine de (keskin dönüş, Omega ilmik, vb.) atamak mümkündür ama bunlar daha ender kullanılır.
DSSP H-bağı tanımları
İkincil yapı hidrojen bağlanması ile tanımlanır, dolayısıyla bir hidrojen bağının kesin tanımı esastır. İkincil yapı için standart H-bağı tanımı, DSSP'nin tanımıdır, bu da tamamen elektrostatik bir modele dayalıdır. Bu model, karbonil karbonu ve oksijene, yükleri, amit azotu ve hidrojene de, yükleri belirler. Elektrostatik enerji değeri şudur:



DSSP'ye göre, eğer ve sadece eğer (ancak ve ancak) -0.5 kcal/mol'den küçük olursa bir H-bağı olabilir. DSSP formülü, fiziksel H-bağı enerjisinin nispeten kaba bir yaklaştırımı olsa da, ikincil yapıyı tanımlamakta genel kabul görmüş bir araçtır.

Protein ikincil yapı öngörüsü
Protein üçüncül yapısını sadece amino asit dizisine dayanarak öngörmek çok zorlu bir problemdir (bakınız protein yapı öngörüsü), ama basit ikincil yapı tanımlamalarını kullanmak daha çözümlenebilir bir problemdir ve uzun zamandır bir araştırma konusu olmuştur.
8-halli DSSP şifresi, bir proteindeki hidrojen bağlanma örüntülerindeki çeşitliliğin bir sadeleştirmesidir. Buna rağmen, ikincil yapı öngörü yöntemlerinin çoğu, daha da ileri giderek, bunları üç ana hâle indirgerler: Sarmal, Yaprak ve Sarım. 8 halden 3 hale dönüşümün nasıl yapıldığı yöntemden yönteme değişir. İkincil yapı öngürüsünün ilk geliştirilen yöntemleri, her bir amino asitin sarmal veya yaprak oluşturma eğilimine dayanıyordu; bazen buna ek olarak, ikincil yapı elemanlarının oluşum serbest enerjisini kestirmek için kurallar da dahi ediliyordu. Bu yöntemler, bir amino asit kalıntısının üç hâlden (sarmal/yaprak/sarım) hangisini benimseyeceğini öngörmekte %60 oranında başarılıydı. Daha sonraları, çoklu dizi hizalamasından yararlanılarak doğruluk oranında önemli bir artış elde edildi (%80'e). Evrimsel olarak birbiriyle ilişkili olan proteinlerin belli bir pozisyonda (ve onun civarında, tipik olarak her iki tarafındaki 7 pozisyonda) yer alabilecek tüm amino asit kalıntılarının dağılımını bilmek, o konum yakınındaki yapısal eğilimler hakkında çok daha iyi bir fikir verir. Örneğin, bir proteinin belli bir konumundan bir glisin olması, orada bir rastgele sarım olduğunu ima edebilir. Ancak, çoklu dizi hizalaması kullanılarak, bir milyar yıllık evrimsel mesafe ile birbirine akraba olan proteinler incelendiğinde, bu proteinlerin %95'inde, o konumda (ve yakın pozisyonlarda) sarmal tercih eden amino asitlerin bulunduğu görülebilir. Üstelik, o ve yakınındaki konumlardaki ortalama hidrofobisite, aynı dizi hizalaması ile incelendiğinde, kalıntıların solvent erişilirliğinin bir α-sarmal yapı ile tutarlılık gösterdiği görülebilir. Bu bilgiler toplu olarak değerlendirildiğinde, söz konusu proteindeki glisinin rastgele sarım değil, α-sarmal bir yapıya ait olması muhtemel bulunur. Mevcut tüm verileri katıştırarak 3-hâlli bir öngörü yapan çeşitli yöntemler vardır, bunların arasında yapay sinir ağları, gizli Markov modelleri ve destek vektör makinaları bulunur. Modern öngörü yöntemleri ayrıca her bir pozisyon için bir güven skoru da verir.
İkincil yapı öngörülerinin karşılaştırmalı olarak değerlendirildiği, örneğin EVA EVA deneyi gibi, çeşitli girişimler vardır. Yaklaşık 270 haftalık bir deneme süreci sonucunda, hâlen en hatasız yöntemler PSIPRED21 Temmuz 2011 tarihinde Wayback Machine sitesinde arşivlendi., SAM, PORTER, PROF ve SABLE olarak belirlenmiştir. İlginçtir, bu farklı yöntemlerin konsensusunu alarak daha iyi bir sonuç çıkmamaktadır. β-yaprakların öngörüsü hâlâ ilerlemeye ihtiyaç gösteren bir konudur; güvenle β-yaprak olduğu öngörülen amino asit kalıntılarının çoğunlukla gerçekten öyle oldukları bulunmuştur, ama bu yöntemler bazı β-yaprakları kaçırmaktadır, yani sahte negatifler vardır. Öngürü doğruluğunun muhtemelen %90 seviyesinde bir üst sınır vardır, DSSP'nin ikincil yapı sınıflarını (sarmal/yaprak/sarım) PDB yapıları ile eşleştirmesinin ayrıntıları nedeniyle.
Hatasız ikincil yapı öngörüsü, çok basit homoloji modelleme vakaları haricinde, üçüncül yapının öngörüsü için temel bir unsurdur. Örneğin, 6 ikincil yapı elemanlı βαββαβ örüntüsü, ferredoksin katlamasının imzası sayılır.
Hizalama
Hem protein hem nükleik asit ikincil yapıları, çoklu dizi hizlamasına yardımcı olmakta kullanılabilir. Sadece basit dizi bilgisi değil, ikincil yapı bilgisi de dahi edilerek bu hizalamaların daha doğru olması sağlanabilir. Bu yaklaşım, RNA için o kadar faydalı olmayabilir çünkü baz eşleşmesi, diziden çok daha fazla korumalıdır. Birincil yapıları hizalanmayacak kadar birbirinden evrimsel olarak ıraksamış proteinlerin ikincil yapılarına bakılarak aralarındaki akrabalıklar bazen fark edilebilir.
Ayrıca bakınız
- Katlanma (kimya)
- Protein birincil yapısı
- Protein üçüncül yapısı
- Protein dördüncül yapısı
- Translasyon
- Yapısal motif
Kaynakça
- Richards F. M., Kundrot C. E. (1988). "Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure". Proteins. 3 (2). ss. 71-84. doi:10.1002/prot.340030202. PMID 3399495.
- Kabsch W., Sander C. (1983). "Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features". Biopolymers. 22 (12). ss. 2577-2637. doi:10.1002/bip.360221211. PMID 6667333. 29 Mayıs 2020 tarihinde Wayback Machine sitesinde arşivlendi.
- Frishman D., Argos P. (1995). "Knowledge-based protein secondary structure assignment". Proteins. 23 (4). ss. 566-579. doi:10.1002/prot.340230412. PMID 8749853.
- [Bottomley] (2004). "Interactive Protein Structure Tutorial". 1 Mart 2011 tarihinde kaynağından arşivlendi. Erişim tarihi: 9 Ocak 2011.
|yazar-bağ1=değerini kontrol edin (yardım)
Daha çok okuma için
- C Branden and J Tooze (1999). Introduction to Protein Structure 2nd ed. Garland Publishing: New York, NY. (İngilizce)
- M. Zuker "Computer prediction of RNA structure", Methods in Enzymology, 180:262-88 (1989). (RNA ikincil yapısını öngörmek için dinamk programlama algoritmaları hakkındaki klasik makale.) (İngilizce)
- L. Pauling and R.B Corey. Configurations of polypeptide chains with favored orientations of the polypeptide around single bonds: Two pleated sheets. Proc. Natl. Acad. Sci. Wash., 37:729-740 (1951). (Beta-yaprak konformasyonu hakkındaki orijinal makale.) (İngilizce)
- L. Pauling, R.B. Corey and H.R. Branson. Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. Wash., 37:205-211 (1951). (alfa- and pi-sarmal konformasyonları hakkında makale; sarmalının mümkün olmadığını göstermiştir.) (İngilizce)

Dış bağlantılar
- NetSurfP - İkincil yapı ve yüzey erişim öngörücüsü 17 Haziran 2011 tarihinde Wayback Machine sitesinde arşivlendi.
- PROF
- Jpred
- PSIPRED21 Temmuz 2011 tarihinde Wayback Machine sitesinde arşivlendi.
- DSSP
- WhatIf
- Mfold
- STRIDE
- PSSpred1 Mayıs 2019 tarihinde Wayback Machine sitesinde arşivlendi.
