Turner sendromu
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.[1][2][3][4]
| Turner sendromu | |
|---|---|
 | |
|
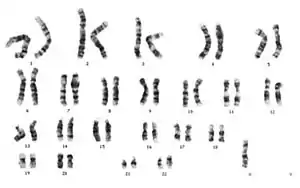
45,X0 karyotipi | |
| Uzmanlık |
Medikal genetik |
Kıkırdaklardaki gelişme geriliği 4 yaşından sonra belirginleşen boy kısalığına neden olur. Yumurtalıkların (ovaryumunlar) oluşumu ve işlevleri yetersizdir ya da hiç görülmez. Östrojen yokluğunun neden olduğu puberte olmaz, primer amenore (adet yokluğu) ve kısırlık saptanır. Dış genital organlarda gelişme kusurları vardır. Puberte hormon tedavisiyle tetiklense bile kısırlık sorunu çok az bir hastada giderilebilmektedir. Ovaryum tümörleri riski yüksektir.[1][2][3][4][5]
Noonan sendromu, Turner sendromu'nun erkek çocuklarında görülen, karyotipin normal olduğu bir sendromdur.
Belirtiler
.jpg.webp)
Kısırlık ve adet yokluğu birincil bulgulardır. Bunlara ek olarak aşağıdaki bulgular bulunabilir:[1][2][3][4]
- Kısa boy (kıkırdaklardaki gelişme geriliği nedeniyle 4 yaşından sonra belirginleşen boy kısalığı)
- Lenfödem (el ve ayaklarda şişkinlik)
- Boyun kısa ve perdeli (pterygium colli), sfenks yüzü izlenimi
- Göz kapakları düşüklüğü (ptozis)
- Üstçenede gelişme geriliği (mikrognati) ve çukur damak
- Altçenede malformasyon
- Çiğneme düzleminde bozulma (maloklüzyon)
- Düşük kulak çizgisi
- Dolaşım sistemi anomalileri (biküspid aort kapağı, aorta koarktasyonu, aorta dilatasyonu, aorta diseksiyonu)[6]
- Kol açısı anomalisi (cubitus valgus)
- Kemiklerde osteoporoz, boyun omurlarında ve patella’da gelişme geriliği
- Deri oluşumları (büyük nevus pigmentosus’lar, molloscum fibrosum), tırnak malformasyonları
- Hipertansiyon
- Böbrek anomalileri
- Hipotiroidizm
- Diabetes mellitus
- vb.
Tarihte
Sendromun adı 1940'larda hastalığı tanımlayan Henry Turner adında bir Oklahomalı endokrinolojist tarafından gelmektedir. Avrupa'da genellikle Ullrich-Turner sendromu ya da Bonnevie-Ulrich-Turner sendromu olarak kullanılmaktadır.
45,X'li karyotipi ik olarak yayımlayan Dr. Charles Ford (1959), Turner sendromunun cinsiyet kromozomlarını ilgilendiren bir eksikliğe bağlı olduğunu keşfetmiştir.
Genetiği
X kromozomu taşımayan bir yumurta hücresinin X kromozomu taşıyan bir sperm ile döllenmesi sonucu ya da eşey kromozomu taşımayan bir spermin normal bir yumurta hücresini döllemesiyle meydana gelir. Eşey kromozomlarından biri dişide bulunmaz ya da yapısı bozulmuş durumdadır. 1/2000 canlı doğan dişi bebekte görülür. Karyotip 45,X ya da 45,XO olarak gösterilir ya da farklı genetik varyasyonları vardır. Bu kromozomal düzensizlik dört farklı yolla meydana gelebilir;
- Mayoz sırasında ayrılmama; eşey kromozomlarından her ikisinin de diğer gruba gitmiş olmasıyla yumurta ya da spermde X ya da Y kromozomunun bulunmaması. Karyotipi 45,X (ya da 45,X0) olarak gösterilir. Bu grup, Turner sendromu içinde %50'lik yer kaplar.
- Gelişme sırasında gametogenezde, X kromozomunun bir parçasını yitirip, diğer ucuna tutunarak halka şeklini almasıyla. Karyotip; 46, XrXp olarak gösterilir. (rXp; halka kromozomun bir parçasının kaybolduğunu gösterir.) Diğer türde ise, X kromozomunun iki uzun kolu birleşir, kısa kol kaybolmuştur. Bu izokromozom olarak adlandırılır. Turner sendromunda %30'luk yer kaplayan gruptur.
- Hücre bölünmesinin erken safhalarında, ayrılmama durumu bir X'in kaybolmasına yol açar. Bu tüm hücrelerde görülmez, bu yüzden mozaizm ortaya çıkar. Karyotip; 46, XX/45, X şeklindedir. Burada mozaizmin yoğunluğuna bağlı olarak hastalığın seyri değişir. Turner sendromunda %20 oranında görülen gruptur.
- Çok nadir olarak, embriyo normal X kromozomuna sahip ve küçük bir Y kromozomu taşıdığında da Turner sendromu bulguları görülür. Burada Y kromozomu fonksiyonel değildir ve bu yüzden kişi Turner sendromlu olarak dişi şeklinde gelişir. Tıpta, bu Swyer sendromu olarak bilinir.
Turner sendromuna yol açan risk faktörleri henüz tam olarak bilinmemektedir. Ayrılmama durumu anne yaşıyla arttığında Down sendromu gibi hastalıklara yol açabilmekteyken, Turner sendromuna herhangi bir etkisi olmamaktadır. Tamamen sağlıklı bir aile de Turner sendromlu bir çocuğa sahip olabilir. Yeni doğan her 2000 kız çocuğunun birinde Turner sendromu görülmektedir.
Oluşum ve teşhis
Yaklaşık olarak Turner sendromlu gebeliklerin %98'i düşükle (spontan abortus) sonuçlanmaktadır. Amerika'da yapılan araştırmaya göre düşüklerin %10'u Turner sendromludur.Canlı doğumlardaki oranı; 1/2000'dir.
Turner sendromu ön tanısı muayene bulgularının yanı sıra bazı hormonal testler ile konabilir. Ancak kesin tanı için kromozom analizi yapılır. Alınan kan örneği ile hücre kültürleri hazırlanır. Kromozomlar incelenerek karyotipleme yapılırarak tehşis konur.
Doğum öncesi tanı
Turner Sendromu gebelikte tanınabilen bir hastalıktır. Bazen fetüs, ultrason bulguları ile de anormal olarak tanımlanabilir. (Kalp bozuklukları, böbrek anomalileri, kistik higroma, gibi). Ultrasonografi, amniyosentez ve diğer bazı tanı yöntemleri ile Turner Sendromundan şüphelenilen gebeliklerde kesin tanı konur. Turner Sendromu saptanmışsa aileye ayrıntılı genetik danışmanlık verilerek gebeliğin sonlandırılması önerilir. Turner Sendromlu Kadınlar Çocuk Sahibi Olabilir mi?
Turner sendromluların bir kısmında ergenlik dönemi başına kadar canlı kalmış az sayıda yumurta hücreleri mevcuttur. Ancak zamanla bu hücreler de hızla azalır. Bu sebeple Turner sendromlu bir kadının “tıbbi yardım” almaksızın çocuk sahibi olma ihtimali son derece düşüktür.
Dış bağlantılar
- Turner Sendromlular Enstitüsü
- Turner Sendromu
- www.ncbi.nlm.nih.gov/Turner sendromu18 Kasım 2004 tarihinde Wayback Machine sitesinde arşivlendi.
- Genetik hastalıklar ve Turner sendromu29 Eylül 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- Medikal Rehber(Türkçe)
- Medikal Ansiklpoedi29 Eylül 2006 tarihinde Wayback Machine sitesinde arşivlendi.
Kaynakça
- Bondy CA. New issues in the diagnosis and management of Turner syndrome. Reviews in Endocrine and Metabolic Disorders, 6(4):269-280, 2005
- Doswell BH, Visootsak J, Brady AN, Graham JM Jr. Turner syndrome: an update and review for the primary pediatrician. Clinical Pediatrics (Philadelphia), 45(4): 301-313, 2006
- Morgan T. Turner syndrome: diagnosis and management. American Family Physician, 76(3):405-410, 2007
- Bondy CA. Turner syndrome 2008. Hormon Research, 71 Suppl 1:52-56, 2009
- Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. European Journal of Endocrinology, 177(3):G1-G70, 2017
- Matura LA, Ho VB, Rosing DR, Bondy CA. Aortic dilatation and dissection in Turner syndrome. Circulation, 116(15):1663-1670, 2007
Ayrıca bakınız
- Genetik hastalık
- Genetik tanı merkezi
- Kalıtsal hastalıklar
- Demir, İ., Genetik, Ege Üniversitesi Ziraat Fakültesi Yayınları
- Demirsoy, A., Yaşamın Temel Kuralları Cilt I Kısım I, Ondokuzuncu basım.
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |
