Bardet-Biedl sendromu
Bardet-Biedl sendromu (Laurence-Moon/Bardet-Biedl sendromu), hipofiz ve hipotalamus kökenli endokrin sistem sendromlarından biridir (bu gruptaki öteki sendromlar: Fröhlich sendromu, Kallmann sendromu, McCune-Albright sendromu, Septo-optic dysplasia). Kalıtsaldır. 24 fenotipi vardır, bunların yalnızca 3’ü önemlidir ve otosomal dominant ya da otosomal resesif (digenik digenik kalıtım17 Mayıs 2018 tarihinde Wayback Machine sitesinde arşivlendi.; iki genin aynı hastalığın kalıtımında etkili olduğu) yolla aktarılır.[2][3][4][5][6]
| Bardet–Biedl sendromu | |
|---|---|
| Diğer adlar | Biedl-Bardet sendromu[1] |
 | |
|
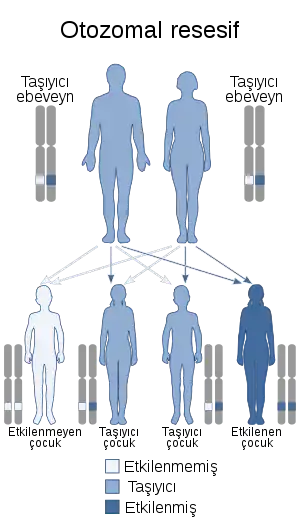
Bu durum otozomal resesif yolla (digenik resesif dahil) kalıtsal olabilir | |
| Uzmanlık |
Medikal genetik |
Brakisefali nitelikleri içeren bir makrofefali saptanır. Alın dardır. Rod-cone distrofisi ve retinitis pigmentosa gibi retina patolojileri ile katarakt gibi patolojiler görme sorunlarına yol açar. Kulak kepçeleri iridir. Basık ve kısa burun kökü altında uzun ve silik bir filtrum vardır. Koku alma bozukluklarından (anosmia) yakınırlar.[2][5][6][7][8]
Ağıziçi bulgular arasında dişeti büyümeleri (gingival hiperplazi), oligodonti ya da hipodonti, küçük dişler (mikrodonti) ve taurodontism önde gelir. Bazı dişlerin kökleri kısadır. Damak kubbesi yüksektir (çukur damak). Radyolojik incelemelerde, çene kemiklerinde yoğunlaşma alanları saptanır (osteoskleroz).[2][5][6][9][10]
Obeziteye eşlik eden diabetes mellitus saptanır. Artı parmaklar (polidaktili) olabilir. Erkeklerde hipogonadizm, kızlarda genital malformasyonlar vardır. Her iki cinsteki progressif böbrek patolojileri (hipoplazi ya da displazi, kistik böbrek, interstisiyel nefrit, böbrek yetmezliği) yaşam kalitesini ve süresini olumsuz etkileyen faktörlerdendir. Öğrenme güçlükleri ve davranış bozuklukları saptanır.[2][5][6]
Kaynakça
- "Bardet-Biedl syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. 28 Ağustos 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 13 Ağustos 2019.
- Beales PL, Elcioglu N, Woolf AS, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. Journal of Medical Genetics, 36(6):437-446, 1999
- Tobin JL, Beales PL. Bardet-Biedl syndrome: beyond the cilium. Pediatric Nephrology, 22(7):926-936, 2007
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. Journal of Clinical Investigation, 119(3):428-437, 2009
- Forsythe E, Beales PL. Bardet-Biedl syndrome. European Journal of Human Genetics, 21:8–13, 2013
- Maksillofasiyal 28 Ocak 2020 tarihinde Wayback Machine sitesinde arşivlendi. sendromlar, Haziran 2020
- Azari AA, Aleman TS, Cideciyan AV, et al. Retinal disease expression in Bardet-Biedl syndrome-1 (BBS1) is a spectrum from maculopathy to retina-wide degeneration. Investigative Ophthalmology & Visual Science, 47:5004–5010, 2006
- Beales PL. Lifting the lid on Pandora's box: the Bardet-Biedl syndrome. Current Opinion in Genetics & Development, 15(3):315-323, 2005
- Andersson E-M, Axelsson S, Gjolstad L-F, Storhaug K. Taurodontism: A minor diagnostic criterion in Laurence-Moon/Bardet-Biedl syndromes, Acta Odontologica Scandinavica, 71(6):1671-1674, 2013
- Haritha A, Jayakumar A. Syndromes as they relate to periodontal disease. Periodontology 2000, 56:65–86, 2011
