Mannosidozis
Mannosidozis (mannosidosis), otosomal resesif yolla aktarılan, mannosidaze enzimi eksikliğiyle karakterize kalıtsal bir lizozomal depo hastalığıdır. Alfa-B ve Beta-A olarak nitelendirilen 2 tipi vardır. Alfa-B tipi olgulardaki bulgular çok sayıdadır ve daha güçlüdür.[1][2]
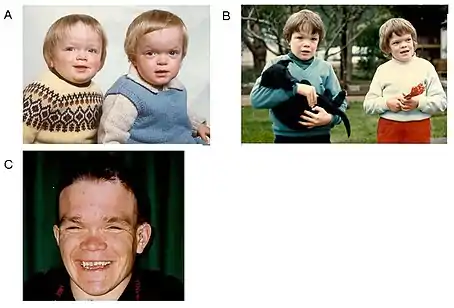
Alfa tip mannosidozis
Fiziksel ve psikomotor gelişme geriliği belirgindir. İri ve kalın kemikli bir kafatası vardır (makrosefali). Artkafa (oksiput) düz, alın geniş ve bombelidir. Hipertrikoz nedeniyle saçlar ve kaşlar gür, saç çizgisi görece aşağıdadır. Göz kapak kıvrımları anomalisi vardır, erişkinlerde retina dejenerasyonu ve nistagmus nedeniyle görme sorunları belirir. Kulaklar iridir, işitme sorunları saptanır.[1][3][4][5]
Yüz yapısı kabadır; üstçene küçük (mikrognati), altçene öne çıkıntılıdır (prognatizm). Dil büyüktür (makroglossi), dişeti büyümeleri belirlenir.[1][3][4][5]
Karaciğer ve dalak büyüktür (hepatosplenomegali).[1][3][4][5]
İskelet sistemi anomalileri oldukça fazladır: Sternum ve kosta anomalileri, disostozis multipleks (madde birikmesine bağlı değişiklikler), çok sayıda vertebra anomalisi ile femurların eğri olduğu görülür.[1][3][4][5]
Sinir sistemiyle ilgili bulgular yaş aldıkça belirginleşir ve giderek ağırlaşır; psikomotor gerilik ve zeka geriliği, hipotoni, konuşma sorunları, ataksi, spastik yapı, reflekslerde aşırılık (hiperrefleksi), erişkin yaşlara doğru beyin ve serebellum atrofisi gelişir.[1][3][4][5]
İmmunoglobulin düzeyi düşüktür; bu nedenle, yineleyen enfeksiyonlar sık görülür. İdrarda bol miktarda mannoz içeren oligosakkarid vardır (oligosakkaridüri).[1][3][4][5]
Beta tip mannosidozis
Alfa-B tipi olgulardakine kıyasla bulgular daha az sayıda ve daha hafiftir. Kafa ve yüz bulguları çok hafiftir. Gözde, konjunktiva damarlarının çok ve belirgin olduğu saptanır. Zeka geriliği, agresyon ve hiperaktif davranışlar izlenir. Hipotoni ve konuşma bozukluklarının yanı sıra periferik nöropatiler vardır. Deride angiokeratoma adı verilen iyi huylu tümörler olabilir. Yineleyen enfeksiyonlar görülür. İdrarda disakkaridler saptanır (disakkaridüri).[6]
Kaynakça
- Borgwardt L, Lund AM, Dali CI. Alpha-mannosidosis - a review of genetic, clinical findings and options of treatment. Pediatric Endocrinology Reviews, 12 Suppl 1:185-191, 2014
- Bedilu R, Nummy KA, Cooper A, et al. Variable clinical presentation of lysosomal beta-mannosidosis in patients with null mutations. Molecular Genetics & Metabolism, 77: 282-290, 2002
- Ölmez A, Nilssen O, Coşkun T, Klenow H. Alpha-mannosidosis and mutational analysis in a Turkish patient. Turkish Journal of Pediatrics, 45:46–50, 2003
- Gutschalk A, Harting I, Cantz M, et al. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology, 63:1744–1746, 2004
- Pittis MG, Montalvo AL, Heikinheimo P, et al. Funtional characterization of four novel MAN2B1 mutations causing juvenile onset alpha-mannosidosis. Clinica Chimica Acta, 375:136–139, 2007
- Wenger DA, Sujansky E, Fennessey PV, Thompson JN. Human beta-mannosidase deficiency. New England Journal of Medicine, 315: 1201-1205, 1986
